
February 2019: Antimicrobial Agents
Sequential non-inferiority studies comparing reducing durations of antibiotics might fail to catch genuine reductions in efficacy. This is just a short letter to the editor, but I think the issue is important enough to merit our attention. Writing of the recent 7 vs 14 days of antibiotics for GNR bacteremia trial by Yahav et al, the authors warn that sequential non-inferiority comparisons between progressively shorter durations of therapy could move the goalposts of what we consider an acceptable outcome for an infection, even without any of the individual studies demonstrating non-inferiority. Basically, it’s the metaphor of the frog slowly boiled in a pot of hot water, but applied to clinical trials.
I’ve sketched up a little diagram to illustrate:
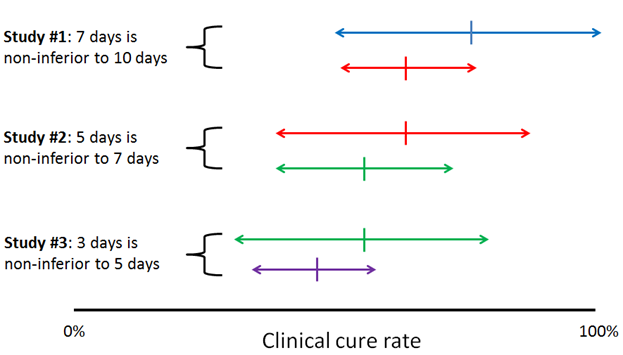
The authors suggest that to combat this, researchers should choose tight non-inferiority margins, choose adequately different durations of therapy (i.e. 2 vs 4 weeks rather than 5 vs 7 days), design multi-arm trials with varying durations to identify a dose-response curve, and retrospectively study populations to identify declining treatment responses as practices shift toward short antibiotic courses.
That all sounds great to me, a person who is not paying for clinical trials’ extra sample sizes. 30726884
Speaking of clinical trial guidelines, 26 experts in infectious diseases, intensive care, and pharmaceutical development published consensus clinical endpoints for future antimicrobial trials in HAP and VAP this month. They included pharma industry representatives in the process, which I understand but don’t love; on the other hand, they used the Delphi method, a great strategy for reaching consensus without discussions yielding to eminence. The clinical endpoint the panel agreed upon for VAP was a hierarchal composite endpoint including survival at day 28, mechanical-ventilation free days through day 28, and clinical cure between days 7 and 10, with the latter defined as the combination of resolution of clinical symptoms of pneumonia as well as stability or improvement of radiologic signs. They had a separate but very similar endpoint for HAP, which I thought was silly. 30722013
You should be side-eying the current minocycline breakpoints for Acinetobacter baumannii. Minocycline, a drug I think of mostly as treatment for S. aureus osteoarticular and soft tissue infections, has recently seen increasing use for A. baumannii. For decades, the CLSI minocycline breakpoints for Acinetobacter have been set at <4 mg/L for susceptible and >16 mg/L for resistant (EUCAST hasn’t issued minocycline breakpoints for A.baumannii). These breakpoints are based on disc diffusion testing and pharmacokinetic versus pharmacodynamics data (as the tetracyclines predate widespread recognition of the importance of pharmacodynamics in predicting antimicrobial efficacy).
The authors review the available PK/PD data for minocycline. Minocycline’s activity is based on AUC/MIC, (specifically, free AUC/MIC, given it is >75% protein bound). They found data indicating that the pharmacodynamic target for minocycline in A.baumannii is an fAUC/MIC of 20-25, and that a 200mg dose of minocycline achieves a fAUC 18-40mg*h/L. Solve for MIC and we find the breakpoint ought to be in the 1-2mg/L range. But wait, that’s not all! Since those data are just means of population distributions, the authors performed a Monte Carlo simulation using published data on variation in achieved minocycline AUCs. Within this model, a 200mg daily dose of minocycline only had an 85% probability of target attainment (fAUC/MIC >25) at an MIC of 0.5mg/L, and even 400mg daily yielded 0% PTA at an MIC of 4mg/L.
If your eyes have glazed over: that’s a lot of pharmacy words for saying that the CLSI breakpoints for minocycline in A. baumannii are probably way too generous, especially given that minocycline’s protein binding is funky and results in diminishing returns with free drug levels when you push the dose. These data suggest the true breakpoint for minocycline at the usual 200mg per day dose should be 0.5mg/L, and would push me to use 200mg twice daily for serious infection with a “susceptible” A. bauumannii with an MIC of 1. 30412249
Chronic bacterial prostatitis? Why not fosfomycin? Unless the prostate is acutely inflamed, most antibiotics don’t achieve great levels in prostatic tissue. As a result, for the longest time the only drugs worth anything for chronic bacterial prostatitis (CBP) were the fluoroquinolones. In fact, if you read the original literature published on prostatitis, you’ll see the the outcomes in the pre-quinolone era literature were universally terrible. Given this, I have my suspicions that the extended course of antimicrobial therapy we give for CBP today is based on the poor responses of this era, and that shorter course therapy with a fluoroquinolone might do just fine - but that’s neither here nor there.
Anyway, guidelines and medical dogma recommend giving 6-12 weeks of a quinolone for CBP. Aside from being a real antimicrobial stewardship faux pas, this practice is becoming infeasible with the spread of quinolone-resistant Enterobacteriaceae in communities worldwide. These organisms are often also resistant to trimethoprim-sulfa and tetracyclines and harbor ESBL genes rendering them resistant to the penicillins and cephalsoporins. So where does that leave us? Well, with fosfomycin – which just so happens to get good levels in the uninflamed prostate.
The authors conducted a prospective observational study of patients with CBP referred to a ID clinic in Athens, Greece over a four-year period. The inclusion criteria were robust: patients had to have >3mo of voiding symptoms or GU/pelvic/rectal pain, a positive urine or expressed prostatic fluid culture, a negative STD PCR panel, a transrectal ultrasound or MRI showing prostatic inflammation, and either resistance to traditional agents (quinolones, TMP/SMX, or minocycline) or a prior treatment failure. The patients received 3g fosfomycin daily for one week following by 3g every other day for 6 weeks, with an optional extension to 12 weeks for patients who had prostatic calcifications on imaging. The primary outcomes of interest were cure (clinical+microbiological) at the end of treatment and relapse at three and six months.
The study included 44 patients; the median age was 54, a third of patients had BPH, and 86% had had a prior episode of CBP. E.coli (66%), K.pneumo (14%), and E.faecalis (14%). Organisms were resistant to quinolones (75%), TMP/SMX (65%), and cephalosporins (23%), but all were susceptible to fosfomycin. Fosfomycin dosing had to be extended to every 72 hours in 4/44 patients and stopped in one patient due to diarrhea; the total incidence of diarrhea in the cohort was 18%, though no cases of C.difficile infection occurred. About half of the patients required extension of therapy to 12 weeks, and the overall rate of cure at the end of therapy was 36/44 (82%). Relapse rates were 20% at 3 months and 23% at 6 months; none of the identified strains harbored fosfomycin resistance, though emergent resistance was detected among the patients who experienced clinical failure on therapy.
In conclusion, extended therapy with fosfomycin appears to be effective and safe – other than causing a lot of diarrhea – for chronic bacterial prostatitis. 30796442
Antabuse might have a new career battling non-tuberculous mycobacteria. This work came out of a microbiology lab at the Central Drug Research Institute in Uttar Pradesh (India). The authors performed drug screening with whole-cell growth inhibition assays to identify new antimycobacterials, followed by cytotoxicity studies in cell culture to establish a selective index. The authors found that disulfiram possessed an MIC of 32 mg/L for the three NTMs they tested, M. fortuitum, M. chelonae, and M. abscessus; in comparison, the 50% cytotoxic concentration was >200mg/L, which could be promising for drug development. They demonstrated that disulfiram exhibits both time and concentration-dependent killing activity, that its in vitro activity at the MIC was similar to amikacin, that it effectively killed intracellular mycobacteria, and that it strongly synergized with fluroquinolones, glycopeptides, and amikacin. In a neutropenic mouse model, 50mg/kg of disulfiram reduced mycobacterial burdens in kidneys by 1.4 log (p<0.001), but did not reduce mycobacterial loads in the spleen.
So, we have some very preliminary evidence to suggest that disulfiram or one of its derivatives might become an additional oral treatment option for NTM infection. I mention “or one of its derivatives” because disulfiram is rapidly metabolized into other substances in people, so the real question is whether said metabolites also possess antimycobacterial activity. Shame about the antabuse effect, but people on TB & NTM meds shouldn’t be drinking anyway. 30753528
While we’re talking mycobacteria, this month CMI published a randomized controlled trial of adding clofazimine to best available therapy for MDR-TB in China. In this case, “best available therapy” meant amikacin or capreomycin, levofloxacin, pyrazinamide, ethambutol, para-aminosalicylic acid, and amoxicillin/clavulanate for six months, followed by another 18 months of fewer drugs. The primary outcome was a combination of “cure” (three consecutive negative sputum cultures by the end of a completed 24mo of therapy) and “treatment completion” (culture conversion by the end of a completed 24mo of therapy, but without three consecutive negative sputum cultures). Over a 2-year period, 74 patients with MDR-TB received the standard of care and 66 received SOC plus clofazimine 100mg daily. At the end of therapy, receipt of clofazimine was associated with a higher rate of favorable outcome (65% vs 47%; RR 1.5; p=0.034). Unfortunately, clofazamine was also associated with a 2-fold high rate of adverse events, most often skin discoloration and hepatitis. To that I say: those side effects are probably still better than having MDR-TB. 30036672